Part of a Texas State University summer math camp I attended in high school included conducting group research projects with Texas State University professors. My research group used R to process and statistically analyze DNA methylation patterns in eight breast cancer cell lines and seven normal samples. DNA methylation plays a crucial role in gene expression as it can interfere with DNA transcription within cells. Specifically, we studied methylation of CG sites (cytosine and guanine pairings) – methylation only occurs on cytosine bases and previous research has shown gene promoters are rich in CG sites in particular. Before directly comparing the breast cancer cell lines to the normal cell lines, we first looked at degrees of variation in DNA methylation within breast cancer cell lines and separately within the normal cell lines. We found that partially methylated sites tended to have a relatively higher standard deviation indicating that differential or heterogenous methylation patterns exist at those CG sites. To narrow down, we selected partially methylated CG sites that had a mean methylation level between 0.4 and 0.6 and standard deviation above or equal to 0.3. These CG sites were then grouped – any CG sites that were within 20 base pairs of another were placed in the same group. We focused on groups with more than 10 CG sites as these were indicative of genes of interest because of high methylation variation within a small portion of a chromosome. We plotted chromosome location and methylation level for each selected group, allowing us to identify specific CpG islands with high methylation variation across cell lines. These CpG islands were marked as potentially influential in breast cancer tumorigenesis.
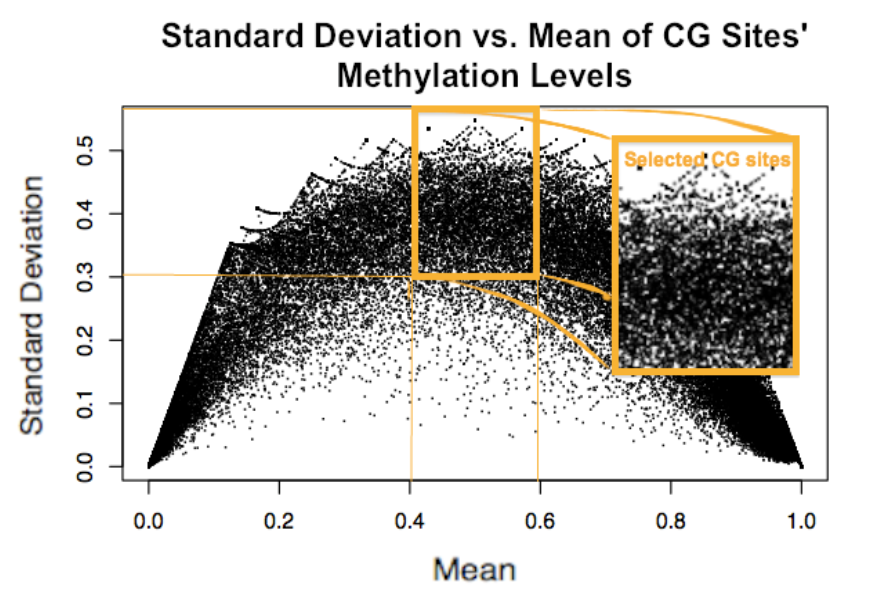
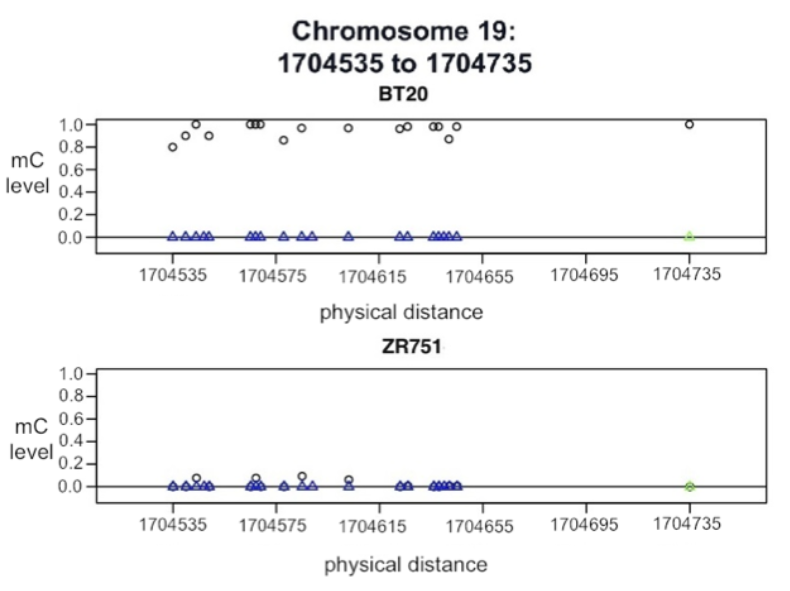
(left to right) Standard deviation vs. mean methylation level of selected CG sites (orange) in breast cancer cell lines; Methylation on chromosome 19, BT20 and ZR751. Blue triangles indicate grouped CG sites, green triangles indicate lone CG sites, and black circles represent methylation levels.
Using the normal cell line data, we computed the standard deviation and mean of the methylation levels at the same CpG islands pinpointed in the breast cancer cell lines. Most CG sites had either no methylation or full methylation, with much smaller standard deviations of less than 0.1. We then used chi-square tests to compare variation levels of the normal data against cancer cell line data based on the hypothesis that the normal cell lines displayed less methylation variation than the cancer cell lines. The CG site groups with at least one hundred significant CG sites that were selected from our chi-square analysis were then mapped to corresponding genes and promoters and compared to entries in known cancer gene databases like PubMeth and GeneCards – in this process, 88 novel genes were found likely to be linked to breast cancer tumorigenesis. To further downselect and validate these genes, we generated a genetic pathway diagram of these 88 genes using ConsensusPathDB software. This software allows us to visualize and understand gene interactions and the impact of methylation errors on directly or indirectly related genes. Based on this genetic pathway diagram, we found 26 out of 88 novel genes were connected in genetic pathways to genes already linked to breast cancer, forming our final list of novel potential breast cancer biomarker genes.
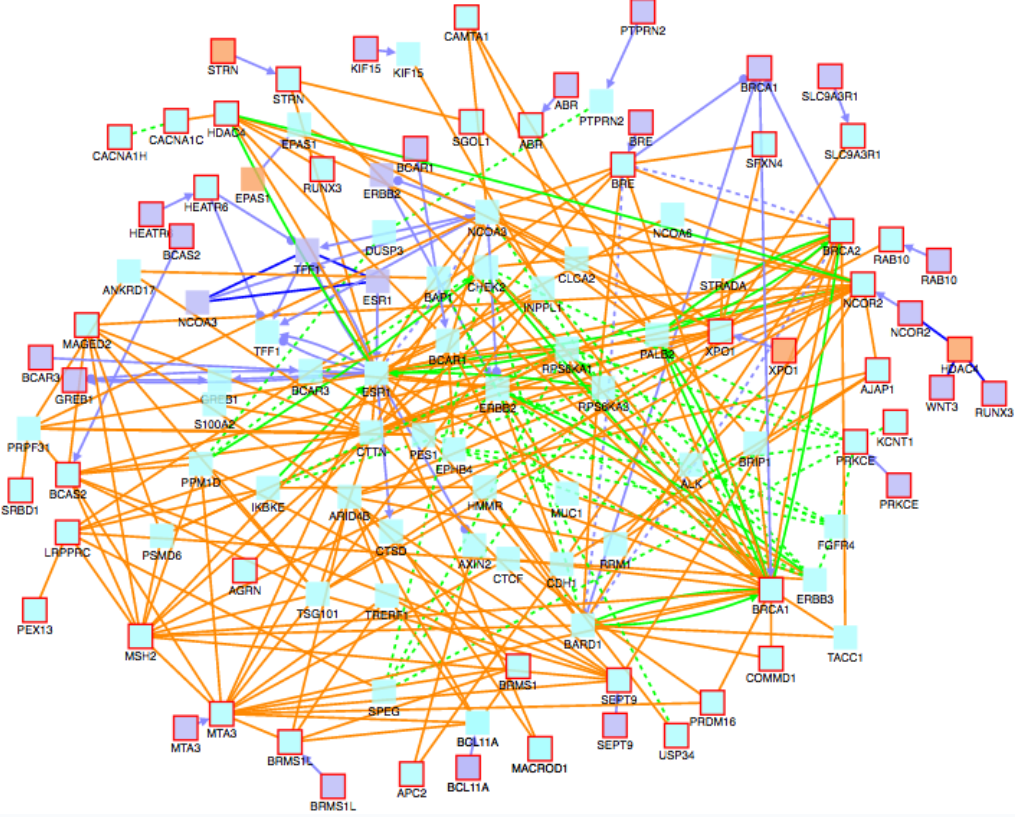
Biomarkers in red linked to genes associated with breast cancer.
In this research, I led the statistical variation and chi-square analyses leading to our initial list of 88 novel genes while other team members led on the genetic pathway analysis. This research led our group to become 2014 Siemens Regional Finalists and was eventually published in the International Journal of Biomedical Data Mining. The experience was my first time understanding the power of statistics and data, how many patterns could lay lurking underneath the surface of huge swathes of data that could lead to significant discoveries and open up new pathways of understanding without ever having to step into a wet lab.
Siemens Regional presentation can be found here, poster presentation here and paper here.